Cowley short range order parameter#
The Cowley short range order parameter can be used to find if an alloy is ordered or not. The order parameter is given by,
where \(n_i\) is the number of atoms of the non reference type among the \(c_i\) atoms in the \(i\)th shell. \(m_A\) is the concentration of the non reference atom.
We can start by importing the necessary modules
[1]:
import pyscal as pc
import pyscal.crystal_structures as pcs
import matplotlib.pyplot as plt
We need a binary alloy structure to calculate the order parameter. We will use the crystal structures modules to do this. Here, we will create a L12 structure.
[2]:
atoms, box = pcs.make_crystal('l12', lattice_constant=4.00, repetitions=[2,2,2])
In order to use the order parameter, we need to have two shells of neighbors around the atom. In order to get two shells of neighbors, we will first estimate a cutoff using the radial distribution function.
[4]:
sys = pc.System()
sys.box = box
sys.atoms = atoms
[5]:
val, dist = sys.calculate_rdf()
We can plot the rdf,
[7]:
plt.plot(dist, val)
plt.xlabel(r"distance $\AA$")
plt.ylabel(r"$g(r)$")
plt.xlim(0, 5)
[7]:
(0.0, 5.0)
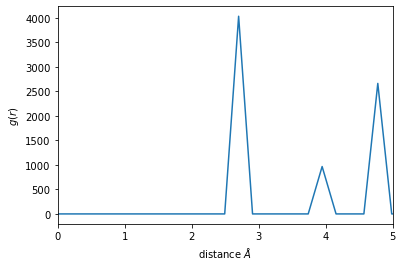
In this case, a cutoff of about 4.5 will make sure that two shells are included. Now the neighbors are calculated using this cutoff.
[8]:
sys.find_neighbors(method='cutoff', cutoff=4.5)
Finally we can calculate the short range order. We will use the reference type as 1 and also specify the average keyword as True. This will allow us to get an average value for the whole simulation box.
[9]:
sys.calculate_sro(reference_type=1, average=True)
[9]:
array([-0.33333333, 1. ])
Value for individual atoms can be accessed by,
[10]:
atoms = sys.atoms
[11]:
atoms[4].sro
[11]:
[-0.33333333333333326, 1.0]
Only atoms of the non reference type will have this value!